Синдром Бругада: самая последняя информация от братьев Бругада, Стетоскоп
Синдром Бругада: самая последняя информация от братьев Бругада
В самом конце августа в JACC опубликован шикарный ориентированный на клинициста обзор о синдроме Бругада, два из авторов которого были братья Brugada. То есть это информация от первоисточника. Ниже даю краткую, но максимально информативную выжимку собственного производства из этой статьи (Josep Brugada, Oscar Campuzano, Elena Arbelo, Georgia Sarquella-Brugada, Ramon Brugada,
Present Status of Brugada Syndrome: JACC State-of-the-Art Review,
Journal of the American College of Cardiology,
Volume 72, Issue 9,
2018,
Pages 1046-1059,
https://doi.org/10.1016/j.jacc.2018.06.037.
(http://www.sciencedirect.com/science/article/pii/S0735109718353622)
Синдром Бругада — генетически обусловленный дефект, проявляющийся своеобразной графикой ЭКГ, с высокой вероятностью ведущий к фибрилляции желудочков и внезапной смерти при структурно не измененном сердце.
В 1992 году впервые этот синдром был описан на основании данных 8 реанимированных пациентов с фибрилляцией желудочков и своеобразной графикой ЭКГ.
Вначале синдром называли синдромом блокады правой ножки пучка Гиса, персистирующей элевации сегмента ST и внезапной смерти.
С 1996 года этот сисндром называется синдромом Бругада.
Вероятно то, что сейчас называется синдромом Бругада, описывалось и ранее. Например в 1917 году на Филлипинах был описан синдром необъяснимой ночной смерти.
В 1998 году обнаружена связь синдрома с генетической аномалией.
Мужчины болеют в 10 раз чаще.
Тестостерон имеет значение: у детей синдром редок, т к уровень тестостерона у девочек и мальчиков близок. После кастрации у мужчин может исчезнуть графика синдрома Бругада.
Оценочная распространенность синдрома 1:2000-5000.
Синдром Бругада ответственен за 4-12% всех внезапных смертей и 20% внезапных смертей при структурно не измененном сердце.
У детей синдром редок, вероятно замаскирован и дезавуируется в более старшем возрасте.
Критерии диагностики:
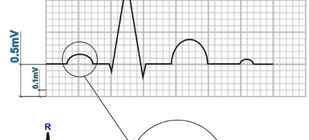
Элевация сегмента ST минимум в одном из отведений V1-V3 на 2 мм и более.
Тип морфологии 1 (см рисунок). Тип 2 не является диагностическим критерием синдрома Бругада, но повышает вероятность его наличия.
Характерная графика может быть зафиксирована на 1 и 2 межреберья выше V1-V3. Рекомендуется в сомнительных случаях записывать ЭКГ не только в стандартных отведениях, но и на 1 и 2 межреберья выше.
Характерная графика может появиться или стать более явной после введения аймалина, прокаинамида или флекаинида.
Графика ЭКГ может быть не вполне типичной.
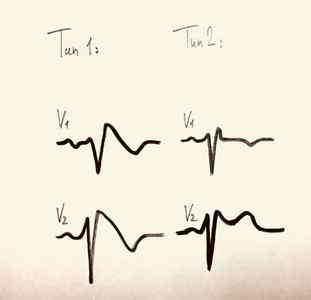
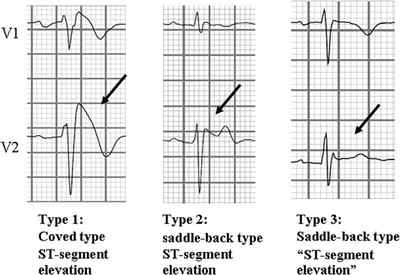
Два типа ЭКГ-графики при синдроме Бругада
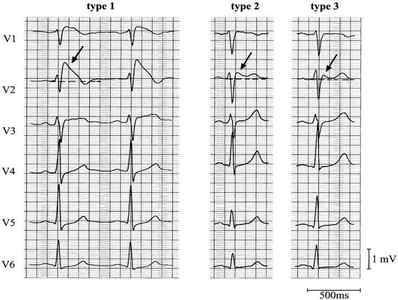
Тип 1 : единственный диагностичный для синдрома. Прямая или изогнутая вверх элевация ST от 2 мм и более, переходящая в отрицательный Т. В минимум одном отведении из V1-V3.
Тип 2 . Не диагностично для синдрома Бругада, но повышает его вероятность и является показанием для фармакологического теста. Седловидная изогнутая вниз элевация ST от 0,5 мм и более. В V1 T может быть любым, в V2-V3 Т позитивный.
Дополнительные критерии при Типе 2:
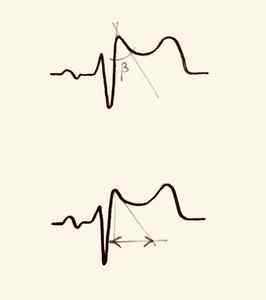
Угол бета на верхнем рисунке равный или превышающий 58 градусов — лучший предиктор трансформации графики Типа 2 в Тип 1 при фармакологическом тесте.
Длина основания треугольника, высота которого 5 мм от точки максимальной элевации сегмента ST. 4 и более мм при скорости ЭКГ 25 мм/с говорит о синдроме Бругада, чувствительность 85%, специфичность 96%.
Клинические проявления:
Синкопе, судорожные приступы, агональное дыхание во сне.
Полиморфная желудочковая тахикардия и фибрилляция желудочков.
Внезапная смерть. Чаще во сне или во время лихорадки. Лихорадка может демаскировать ЭКГ-признаки.
Средний возраст наступления внезапной смерти 41+/- 15 лет.
Фармакологические пробы:
Показания: любое подозрение на синдром (например синкопе или ФЖ), графика синдрома Бругада типа 2.
Тест позитивен, если появляется графика типа 1.
Используют в/в введение аймалина, прокаинамида или флекаинида. При недоступности можно использовать пропафенон или флекаинид per os.
Тест прекратить, если появились частые желудочковые экстрасистолы и более сложные аритмии, а так же при расширении QRS более 130% от изначальной длительности.
25% всех тестов ложноотрицательны. Целесообразно повторить тест с разными препаратами.
Дозы препаратов не описаны в обзоре.
Насколько тесты опасны, не ясно.
Кого лечить? Стратификация риска
Очевидный фактор риска — синкопе, обусловленное желудочковой аритмией.
При отсутствии симптомов четких рекомендаций нет. Лечение индивидуализировано. При позитивном ЭФИ можно рассмотреть вопрос установки кардиовертера.
Кардиовертер — самый надежный способ лечения.
У некоторых пациентов проводят эпикардиальную абляцию, но долговременных результатов нет и эффект не ясен.
http://phonocardio.com/brugada/
Понятие, лечение и признаки синдрома Бругада на ЭКГ
Синдром Бругада – практически не изученная сердечная патология, приводящая к быстрому и непредсказуемому летальному исходу. Она часто не диагностируется даже после смерти, поэтому остается одним из самых загадочных заболеваний.
Что представляет собой синдром?
Синдром Бругада – патологическое состояние с не до конца изученным патогенезом. Открыли его испанские врачи-кардиологи братья П. и Д. Бругада в 1992 году.
Главным открытием ученых было установление причины разнообразных патологий сердечного ритма, из-за которых может наступить внезапная остановка сердца. Это связано с нарушением ионной мембранной проницаемости сердечной мышцы, вследствие чего нарушается проводимость электрических импульсов, которые задают сердечный ритм. Возникшие нарушения могут привести к фибрилляции предсердий, которая часто заканчивается летально.
Ученые заметили, что это заболевание имеет наследственный характер, и предположили генетическое происхождение заболевание. В ходе изучения патологии оказалось, что они были правы – в основе заболевания лежит генная мутация. Было обнаружено несколько генов, поражение и изменение которых привело к развитию заболевания.
Предполагают аутосомно-доминантный тип наследования. Из этого следует, что передача болезни не зависит от пола ребенка и для ее развития достаточно одного дефектного гена. Частота встречаемости этого синдрома 1:10 000. Наиболее подверженными ему являются представители азиатских народов. Пик развития опасных состояний – 30-50 лет, заболевание поражает больше мужчин, с частотой в 8 раз выше, чем женщин.

Причины развития, патогенез
По неизученным причинам происходит генная мутация в отвечающей за работу ионных каналов хромосоме. Из-за этого возникают нарушения функционирования натриевых и кальциевых каналов сердечной мышцы. Нормальная их работа обеспечивает правильную сократительную и проводящую деятельность сердца, то есть регулирует сердечный ритм. Но из-за изменения этих генов, возможны самые разнообразные нарушения ритма сердца.
Выявлено 6 генов, изменение в строении которых приводит к возникновению синдрома. Помимо этого, изучают еще несколько генов, предполагая, что и их мутация способствует развитию болезни. На основании изученных изменений ученые классифицировали заболевание в зависимости от дефектного гена таким образом:
- BrS-1 – мутирующий ген обнаружили в третьей хромосоме, нарушения в которой приводит к патологии проводимости ионов в натриевых каналах. Это самый распространенный вариант заболевания. Его носители подвержены синдрому слабости синусового узла, фибрилляции предсердий и другим опасным нарушениям ритма;
- BrS-2 – патологический ген находится на третьей хромосоме. Блокируется компонент, необходимый для правильного функционирования натриевых каналов;
- BrS-3 – дефектный ген обнаруживается на 12-й хромосоме. Это приводит к нарушению проводимости кальциевых каналов L-типа;
- BrS-4 – генетическая патология на 10-й хромосоме. Проявления аналогичны BrS-3 типу заболевания;
- BrS-5 – локализация пораженного гена на 11-й хромосоме. Распространенный вариант болезни. Кодирует один из протеинов натриевых каналов;
- BrS-6 – обусловлено заболевание нарушениями на 19-й хромосоме. Проявляется аналогично первому варианту заболевания.
В основе патогенеза лежит нарушение трансмембранного потенциала из-за нарушения проводимости ионов натрия и кальция в кардиомиоциты. Это приводит к возникновению нарушения возбудимости, передачи возбуждения в другие ткани, нарушению сократительной способности сердца. Из-за описанных нарушений возникают различные блокады проводящих путей, аритмии, тахикардии, фибрилляции и другие сбои в работе сердца. В зависимости от объема поражения будет меняться клиническая картина. Чем больше нарушений – тем больше и симптоматика.
Ученые еще не выяснили, как именно происходит наследование недуга. Неясным остается, почему поражаются в основном мужчины. Предполагают, что заболевание имеет доминантный тип наследования, но, чтобы подтвердить или опровергнуть это, слишком мало информации.
Симптомы и диагностика патологии
Опасность болезни заключается в отсутствии ярко выраженной симптоматики. Часто первые признаки обнаруживаются случайно, во время планового обследования, на ЭКГ.
Основные проявления
Возраст обнаружения первых изменений на кардиограмме варьируется от 2-3 лет, до 40-50 лет. После выявления нарушений на ЭКГ, возможно отсутствие симптомов следующие 10-15 лет.
Наиболее выраженная симптоматика, возникает в возрасте 35-50 лет. Именно в этом возрасте носители гена обращаются за медицинской помощью. Пациенты предъявляют жалобы на частые приступы тахикардии, головные боли, которые часто имеют мигренозный характер, сильные головокружения, переходящие в обмороки. Данные признаки заболевания преобладают в ночное время, что говорит о влиянии на заболевание парасимпатической нервной системы.
В некоторых случаях проявления заболевания усиливаются после приема некоторых медикаментов: противоаллергических препаратов, бета-адреноблокаторов, ваготонических средств. Из-за отсутствия других специфических симптомов больные редко обращаются к кардиологам. Несмотря на редкое проявление заболевания, довольно высоким остается риск развития внезапной остановки сердца.
Расшифровка ЭКГ
При появлении любой сердечной симптоматики необходимо проверять работу органа при помощи ЭКГ. Взрослым людям рекомендуется проходить обследование планово, без наличия симптоматики. С возрастом частота обследования работы сердечной мышцы увеличивается. Именно при плановых обследованиях чаще всего и выявляют патологические изменения. При наличии синдрома Бругада на ЭКГ возможны следующие нарушения:
- подъем (элевация) сегмента ST над изолинией;
- зубец Т отрицателен в V1-V3;
- признаки блокады ножек пучка Гиса;
- при применении Холтеровского мониторинга обнаруживаются приступы параксизмальной тахикардии либо фибрилляции предсердий.
Рекомендуется пройти генетическое обследование на наличие данного синдрома людям с внезапно возникающими нарушениями и перебоями в работе сердца. Особенно в диагностировании нуждаются молодые люди с описанной выше симптоматикой.
Из-за неопределенной этиологии заболевания не удалось разработать патогенетическую терапию. Не существует консервативных способов предупредить возникновение приступа. Вся терапия данной патологии сводится к симптоматическому лечению, в том числе и неотложным реанимационным мероприятиям. В зависимости от степени клинических проявлений применяют медикаментозные и хирургические методы лечения:
Консервативная терапия
Для купирования возникших приступов аритмии применяются антиаритмичные препараты. Наиболее эффективными является антиаритметик III класса Амиодарон. В некоторых случаях для профилактики внезапной смерти пациента врачи назначают в небольших дозах Хинидин, Дилтиазем, Бретилиум. Но особой эффективности от данных препаратов врачи не замечают.
Следует учесть, что при этом заболевании очень широкий спектр противопоказанных препаратов, влияющих на ритм сердца. Средства, применяемые при аритмиях и тахикардиях, могут ухудшить состояние либо спровоцировать сердечный приступ. Это связано с тем, что механизм действия подобных средств заключается в блокировании натриевых и кальциевых каналов. Такими же являются и признаки заболевания. То есть медикаменты активируют те процессы, которые необходимо остановить.
Оперативная методика
Несмотря на широкий спектр препаратов, улучшающих ритм сердца, фармакологи не изобрели средство, способное предупреждать возникновение приступов и останавливать начавшийся эпизод нарушения ритма, в том числе и фибрилляцию предсердий и асистолию.
Эффективнее всего контролировать болезнь удается при помощи внедрения в сердце искусственного водителя ритма – кардиостимулятора. Аппарат сам создает необходимые в данный момент электрические импульсы, которые возбуждают сердечную мышцу, в результате она начинает правильно сокращаться.
Современные кардиостимуляторы укомплектованы дефибрилляторами, которые в момент возникновения опасного состояния немедленно его купируют. Благодаря работе стимулятора, пациенты могут и не догадываться, что буквально несколько минут назад они были при смерти. Но такое оперативное вмешательство требует придерживаться определенных правил в повседневной жизни. О них пациента обязательно информирует врач перед процедурой.
Открытие синдрома Бругада является огромным прорывом в современной медицине. После его диагностирования врачам удается успешно предотвращать возникновение опасных состояний, используя оперативные методы лечения. Главное – не игнорировать внезапные сбои в работе сердца, а обязательно обратиться за медицинской помощью. Только в таком случае удастся выявить синдром и предотвратить осложнения.
http://simptomov.com/kardio/sindrom-brugada/
Синдром Бругада (СБ): понятие, причины, проявления, диагностика, как лечить
Синдром Бругада (СБ) представляет собой наследственную патологию, связанную с высоким риском внезапной смерти по причине аритмий. Страдают им преимущественно молодые люди, чаще – мужского пола. Впервые о заболевании заговорили в конце прошлого столетия, когда испанские врачи, братья П. и Д. Бругада, описали это состояние и сформулировали основной характеризующий его ЭКГ-феномен.
К проблеме внезапной сердечной смерти внимание врачей приковано давно, но объяснить ее далеко не всегда возможно. Если при хронической ишемической болезни сердца, инфаркте все более или менее ясно, в сердце происходят известные изменения, есть субстрат для появления аритмий, в том числе, смертельно опасных, то во многих других случаях, особенно среди молодых пациентов, вопрос внезапной гибели остается неразрешенным.
Многочисленные исследования и возможности современной медицины позволили отыскать некоторые механизмы внезапных аритмий и остановки сердца у, казалось бы, абсолютно здоровых людей. Известно, что такая патология может носить генетический характер, а, значит, риску подвержены не только носители гена, страдающие нарушениями сердечного ритма, но и их не обследованные родственники.
Низкий уровень выявляемости синдромов, сопровождающихся внезапной смертью в молодом возрасте, недостаточное внимание со стороны врачей поликлиник приводят к тому, что правильный диагноз зачастую не ставится даже после смерти. Малое количество информации об особенностях патологии и отсутствие каких-либо структурных нарушений в миокарде и сердечных сосудах «выливаются» в довольно неопределенные заключения наподобие «острой сердечной недостаточности», причину которой объяснить никто не в состоянии.
Синдром Бругада в числе других состояний, сопровождающихся внезапной гибелью пациентов, — самое «загадочное» заболевание, данных о котором в отечественной литературе практически нет. Описаны единичные случаи патологии, но и среди них не все имеют достаточный объем сведений об особенностях ее течения. Мировая статистика же свидетельствует о том, что более половины всех аритмогенных смертей, не связанных с поражением миокарда и коронарных сосудов, приходится именно на СБ.
Точные цифры распространенности СБ отсутствуют, но исследования уже показали, что среди больных преобладают жители Юго-Восточной Азии, Кавказа, Дальневосточного региона. Высока частота внезапной ночной гибели в Японии, На Филиппинах, в Таиланде. Афроамериканцы, напротив, не страдают этим типом нарушения сердечной деятельности, что, вероятно, связано с генетическими особенностями.
Причины и механизмы развития синдрома Бругада
Среди причин синдрома Бругада указывают генетические аномалии. Было замечено, что патология больше распространена среди членов одной семьи, что стало поводом для поиска генов, ответственных за патологию ритма сердца. Уже описаны пять генов, которые могут быть причиной аритмии и остановки сердца.
Педро Бругада — соавтор формулировки синдрома-эпонима
Установлен аутосомно-доминантный вариант передачи синдрома Бругада, и «виновником» всему считается ген SCN5a, находящийся в третьей хромосоме. Мутации этого же гена зарегистрированы и у больных с иными формами нарушения проведения импульсов в миокарде с высокой вероятностью внезапной смерти.
В кардиомиоцитах, составляющих сердечную мышцу, происходят многочисленные биохимические реакции, связанные с проникновением внутрь и выведением наружу ионов калия, магния, кальция, натрия. Эти механизмы обеспечивают сократимость, правильный ответ на поступление импульса по проводящей системе сердца. При синдроме Бругада страдают белки натриевого канала клеток сердца, следствием чего становятся нарушение восприятия электрических импульсов, повторное «вхождение» волны возбуждения в миокард и развитие аритмии, которая грозит остановкой сердца.
Признаки нарушения сердечной деятельности обычно возникают в ночное время или во время сна, что связывают с физиологическим преобладанием парасимпатической нервной системы, снижением частоты сокращений сердца и интенсивности проведения импульсов во сне.
Клинические и электрокардиографические особенности синдрома Бругада
Симптомы синдрома Бругада малочисленны и очень неспецифичны, поэтому по клиническим характеристикам диагноз можно лишь предположить. Особого внимания заслуживают больные с ниже перечисленными явлениями, в семье которых уже случалась необъяснимая гибель родственников молодого возраста во сне.
Среди признаков синдрома Бругада отмечаются:
- Частые обморочные состояния;
- Приступы сердцебиения;
- Удушье ночью;
- Эпизоды срабатывания дефибриллятора во сне;
- Внезапная некоронарогенная остановка сердца, преимущественно ночью.
Обычно заболевание проявляется у людей среднего возраста, около 40 лет, но описаны случаи патологии и среди детей, а также начало приступов аритмии и потери сознания в пожилом и даже старческом возрасте. Внезапная гибель более чем в 90% случаев наступает, когда больной спит, чаще — во второй половине ночи, что вызвано преобладанием тонуса парасимпатики в это время суток. К слову, у больных хронической ишемией сердца и инфарктом подобные фатальные осложнения чаще регистрируются ранним утром.
Электрокардиографические изменения являются важным диагностическим критерием синдрома Бругада и неотъемлемой частью проявлений, без которой заподозрить патологию невозможно, поэтому ЭКГ должна быть обязательно проведена всем пациентам, предъявляющим жалобы на перебои в ритме сердца и обморочные состояния.
ЭКГ-признаки синдрома Бругада:
ЭКГ-признаки различных типов синдрома Бругада
Желудочковая тахикардия и фибрилляция — самые частые причины внезапной гибели больного , а установка дефибриллятора способна помочь пациенту избежать их, поэтому проблема профилактики синдрома Бругада требует определения вероятности остановки сердца при таких аритмиях. Среди факторов, которые оцениваются для каждого пациента, важны наследственность, эпизоды синкопальных состояний (обмороков), характерные ЭКГ-феномены, особенно, в комбинации с обмороками, результаты холтеровского мониторирования, выявление мутировавших генов.
Для диагностики синдрома Бругада важно тщательно выяснить симптоматику, наличие случаев внезапной необъяснимой смерти среди молодых родственников. Большой объем информации дает динамический ЭКГ-контроль, а также электрофизиологическое исследование сердца с применением фармакологических проб.
Лечение синдрома Бругада
Лечение синдрома Бругада активно обсуждается, специалисты предлагают подходы в назначении лекарственных препаратов, основанные на клиническом опыте и результатах их применения больными с патологией электрической активности сердца, но по сей день так и не найдено эффективного медикаментозного способа профилактики желудочковых аритмий и внезапной смерти.
Больные, у которых ЭКГ-феномены провоцируются пробами с введением блокатора каналов натрия, но при этом симптоматика в покое отсутствует, а в семье не зарегистрировано случаев внезапной гибели, нуждаются в наблюдении.
Медикаментозная терапия состоит в назначении антиаритмических средств класса IА – хинидина, амиодарона, дизопирамида. Стоит отметить, что препараты новокаинамид, аймалин, флекаинид, относящиеся к I классу, вызывают блокаду натриевых каналов и, соответственно, симптоматику синдрома Бругада, поэтому их следует избегать. Провоцируют аритмию, а потому противопоказаны флекаинид, прокаинамид, пропафенон.
Хинидин обычно назначается в небольших дозировках (300-600 мг), способен предупреждать эпизоды желудочковой тахикардии, может применяться у пациентов с разряженным дефибриллятором как дополнительное средство профилактики внезапной смерти.
Эффективным считается изопротеренол, действующий на бета-адренорецепторы сердца, который можно комбинировать с хинидином. Этот препарат может способствовать снижению сегмента ST к изолинии и применим в педиатрической практике. Новым препаратом, «возвращающим» сегмент ST в нормальное положение, является фосфодиэстераза.
Показано, что многие антиаритмические средства вызывают блокаду каналов натрия в кардиомиоцитах, поэтому логично было бы предположить, что более безопасными будут те, которые не имеют такого эффекта – дилтиазем, бретилиум, но исследования их эффективности еще не проводились.
Антиаритмическая терапия оказывается эффективной только у 60% пациентов, у остальных не удается достичь безопасного состояния только при помощи лекарственных препаратов, и возникает необходимость в коррекции электрической активности сердца с помощью специальных приборов.
Самым эффективным способом профилактики внезапной смерти считают установку кардиовертера-дефибриллятора, которая нужна, если:
- Есть симптоматика СБ;
- Течение патологии бессимптомное, но провокация вызывает фибрилляцию желудочков;
- При пробах возникает феномен Бругада тип 1, а среди родственников имелись случаи необъяснимой гибели в молодом возрасте.
По данным мировой статистики, СБ встречается куда чаще, нежели фигурирует в диагнозах кардиологов. Малый процент выявляемости может быть объяснен недостаточной настороженностью в его отношении со стороны врачей, отсутствием убедительных диагностических критериев. Исходя из этого, все больные, имеющие характерные ЭКГ-изменения, необъяснимые обмороки, неблагополучный семейный анамнез в отношении внезапной смерти среди молодых, нуждаются в тщательном обследовании с проведением ЭКГ, холтеровского мониторирования, фармакологических проб. Повышенного внимания требуют и родственники в семьях, где уже были случаи внезапной гибели молодых лиц.
Исследование синдрома Бругада продолжается, а для получения высоких результатов необходимо достаточное количество наблюдений, поэтому специалисты заинтересованы в выявлении как можно большего числа больных в разных странах.
Для изучения патологии создан специальный Международный фонд синдрома Бругада, где могут быть проконсультированы бесплатно и заочно все лица с подозрением на это заболевание. Если диагноз подтвердится, то больного внесут в единый список пациентов, которые в будущем могут быть подвергнуты генетическим исследованиям в целях уточнения наследственных механизмов развития патологии.
http://sosudinfo.ru/serdce/sindrom-brugada/
Проявления и лечение синдрома Бругада
Синдром Бругада – генетическая аномалия, приводящая к нарушению ритма сокращений сердца. Точная распространенность заболевания неизвестна. Это связано с трудностью диагностики патологии, так как недуг может не проявляться клинически. Врачи предполагают, что синдром Бругада занимает лидирующую позицию среди причин внезапной гибели молодых пациентов. Лечение заболевания основано как на применении медикаментозных средств, так и на проведении операции по установке дефибриллятора.
Причины и классификация синдрома Бругада
Известно, что патология обладает наследственной природой. Согласно последним имеющимся сведениям, существует как минимум 6 генов, мутации которых провоцируют возникновение специфических признаков. На основании этой дифференциации в небольшом списке литературы по синдрому Бругада описано несколько вариантов заболевания. Классификация выглядит следующим образом:
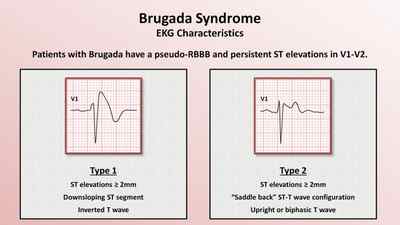
ЭКГ-признаки синдрома
Основные признаки патологии
Клиническая картина заболевания зачастую неспецифична. Этот факт значительно осложняет процесс диагностики недуга. В большинстве случаев признаки синдрома Бругада сводятся лишь к обморокам, а также приступам учащенного сердцебиения в ночные часы. В литературе описаны и случаи, когда заболевание являлось случайной находкой у клинически здоровых пациентов. По этой причине исследователи связывают многие внезапные смерти, возникающие вследствие нарушения ритма работы кардиальных структур, с данным генетическим заболеванием. В статьях, описывающих синдром Бругада, которые можно найти в УДК, подробно описаны лишь критерии постановки диагноза по результатам ЭКГ. Поэтому симптомы поражения зачастую не используются для подтверждения наличия проблемы. Кроме общей слабости, синкопе и приступов тахикардии, при отсутствии физических нагрузок пациенты страдают также от аномальных реакций на некоторые лекарственные средства, например, на антигистаминные препараты и бета-адреноблокаторы. Клинические признаки патологии чаще всего отмечаются в возрасте 30–40 лет, однако в литературных источниках есть и данные о выявлении недуга у детей.
Диагностические исследования
Подтверждение наличия заболевания – важный вопрос в современной медицине. Трудности с выявлением проблемы связаны с тем фактом, что она редко проявляется, а лишь провоцирует внезапную смерть. С целью предотвращения подобных последствий возникновения генетического недуга разработаны диагностические критерии, подразумевающие подробное описание результатов электрокардиограммы при синдроме Бругада. Данный метод считается основным способом подтверждения наличия заболевания, поскольку только с его помощью врачам удается зафиксировать специфические отклонения в работе сердца. При сравнении ЭКГ здорового человека и пациента с врожденным нарушением проведения нервного импульса отмечают возникновение следующих признаков:
Для постановки диагноза потребуется также тщательный сбор анамнеза. Это связано с наследственной природой синдрома Бругада. У пациентов, в семье которых уже зафиксированы случаи внезапной гибели, врачам следует уделять особое внимание работе сердца. Подтверждение наличия патологии подразумевает и проведение генетических тестов, которые позволяют выявить мутации участков ДНК. С целью оценки структуры сердца используется УЗИ, позволяющее сделать специфические фото органа. По снимкам проводятся замеры, а также осуществляется оценка сократительной функции.
Борьба с поражением значительно затруднена. Это связано с отсутствием адекватной и своевременной диагностики патологии. При этом лечить пациентов можно как при помощи медикаментозных средств, так и с использованием хирургических техник, подразумевающих установку кардиостимулятора. При этом консервативные методы значительно уступают радикальным по эффективности.
Лекарственная терапия
Далеко не все антиаритмические препараты могут быть использованы у пациентов с генетической аномалией. Это связано с различным механизмом действия этих медикаментов. Например, лечение при помощи блокаторов натриевых каналов при синдроме Бругада способно привести к ухудшению состояния пациента. При данной патологии применяются такие препараты, как «Хинидин» и «Дизопирамид». Они демонстрируют хорошие результаты при борьбе с приступами пароксизмальной тахикардии. При этом ответ на медикаментозное лечение отмечается лишь у 60% пациентов.
Установка дефибриллятора
Имплантация прибора на сегодняшний день считается самым эффективным способом лечения синдрома Бругада. Она необходима при появлении клинических признаков заболевания, выявлении фибрилляции в ходе холтеровского мониторирования, а также при положительной пробе с использованием блокаторов натриевых каналов. Кардиовертер-дефибриллятор позволяет предотвратить внезапную гибель пациента за счет коррекции ритма сердечных сокращений.
Прогноз и рекомендации по профилактике
Исход патологии определяется интенсивностью ее клинических проявлений. Если у больного отмечается лишь наличие специфических признаков на электрокардиограмме, прогноз благоприятный, особенно при своевременном лечении. Без дефибриллятора велик риск внезапной остановки сердца.
Существуют исследования, которые указывают на мультифакторную природу заболевания. Врачи склоняются к тому, что на интенсивность клинических признаков поражения влияет не только вид генетической мутации, который привел к возникновению проблемы, но и экологическая обстановка, а также гормональный фон в организме человека и образ его жизни.
Фенотипические проявления используются при прогнозировании исхода заболевания и ответа на лечение. Доказано, что в группе риска относительно фатальных осложнений синдрома Бругада пациенты с постоянно повторяющимися обмороками, агональным дыханием на фоне пароксизмальной тахикардии в ночные часы, а также с судорогами неясной этиологии. Таким больным врачи рекомендуют установку имплантируемого кардиовертер-дефибриллятора, который позволяет снизить шанс развития внезапной смерти.
При этом до сих пор ведутся споры по поводу оправданности использования прибора у пациентов, которые в повседневной жизни не сталкиваются с клиническими проявлениями синдрома Бругада.
Ряд докторов склоняется к тому, что при наличии специфической картины на ЭКГ пациентам требуется проведение операции. Другие утверждают, что имплантация оправдана только при проявлении симптомов поражения.
Профилактика развития синдрома Бругада не разработана. Предупреждение возникновения проблемы сводится к кариотипированию родителей на этапе планирования беременности. Для предотвращения формирования фатальных осложнений важно вовремя диагностировать имеющуюся проблему.
Евгений, 29 лет, г. Казань
Обратился к врачу с жалобами на приступы усиленного сердцебиения по ночам. Кардиолог меня осмотрел, послушал, отправил на ЭКГ. В результате выявили признаки, характерные для генетической патологии, – синдрома Бругада. Врач выписал антиаритмические препараты, рекомендовал регулярно приходить на осмотр. Велика вероятность того, что потребуется операция по установке дефибриллятора.
Екатерина, 34 года, г. Томск
История моей семьи богата сердечными заболеваниями, поэтому регулярно посещаю кардиолога. На ЭКГ врач увидел отклонения, которые свидетельствуют о наличии проблемы. По результатам обследования и генетического теста поставили диагноз «синдром Бругада». Пью «Хинидин», регулярно показываюсь кардиологу. Врач говорит, что пока предпринимать радикальные действия смысла нет.
http://prosindrom.ru/genetic/sindrom-brugada.html
Типы экг синдрома бругада
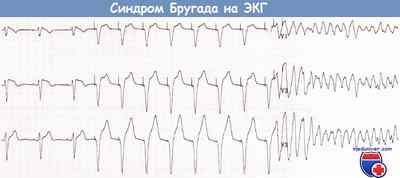
Синдром Бругада характеризуется наличием типичной ЭКГ-картины в виде косонисходящего подъема сегмента ST в отведениях V1, V2 и иногда V3 (как правило, вместе с неполной блокадой ПНПГ), отсутствием структурных изменений со стороны сердца и повышенным риском внезапной смерти от ФЖ или развитием синкопальных состояний вследствие полиморфной ЖТ. Распространенность составляет 1:5000.
Диагностика синдрома Бругада
Наиболее типичные изменения желудочкового комплекса обычно выявляются в отведениях V1 и V2 (I тип): желудочковый комплекс заканчивается положительным отклонением с амплитудой >2 мм (наподобие зубца J, наблюдаемого при гипотермии), за которым следует косонисходящий сегмент ST и отрицательный зубец Т. Интервал PR бывает удлинен. Пароксизмы ФП не являются редкостью. Часто выявляются поздние потенциалы.
У некоторых пациентов типичные ЭКГ-признаки бывают интермиттирующими. Временами подъем сегмента ST может принимать вогнутую или седловидную форму (II или III тип). Такого рода изменения сами по себе не являются достаточными диагностическими признаками синдрома Бругада. Кроме того, ЭКГ может становиться нормальной.
Диагностическое значение ЭКГ можно повысить, разместив грудные отведения на одно или два межреберья выше обычной позиции. Это следует иметь в виду при лечении пациентов, госпитализированных по поводу синкопального состояния неясного происхождения или реанимированных в связи с необъяснимой ФЖ.
С диагностической целью можно применить пробу с внутривенным введением аймалина (1 мг/кг за 5 мин) или, при его недоступности, флекаинида (2 мг/кг за 10 мин). При наличии на ЭКГ признаков, позволяющих предположить диагноз синдрома Бругада, введение данных препаратов приводит к появлению типичных ЭКГ-признаков синдрома Бругада I типа. Иногда ЭКГ-признаки появляются или усугубляются на фоне лихорадки.
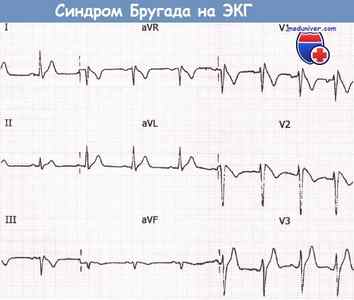
Рутинная ЭКГ с признаками синдрома Бругада, зарегистрированная у медицинского работника, который впоследствии внезапно скончался.
Причины синдрома Бругада
Причиной синдрома Бругада являются генетически обусловленные нарушения функции натриевых ионных каналов. Описано несколько генетических аномалий, связанных с этим синдромом. Не у всех пациентов в семейном анамнезе выявляются случаи внезапной сердечной смерти. Бывают случаи, обусловленные мутацией.
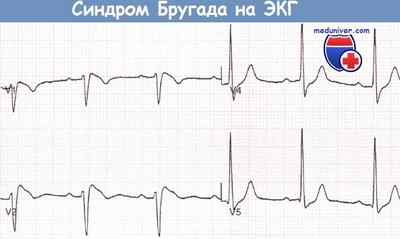
Запись грудных отведений ЭКГ пациента с синдромом Бругада, реанимированного после фибрилляции желудочков (ФЖ), развившейся во время управления автомобилем.
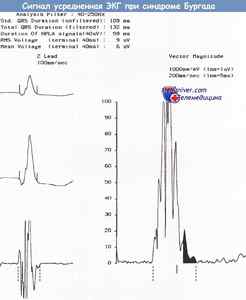
Сигнал-усредненная ЭКГ, зарегистрированная у того же пациента водителя автомобиля, что и на рисунке выше, демонстрирует наличие поздних потенциалов.
Фибрилляция желудочков при синдроме Бругада
Фибрилляция желудочков при синдроме Бругада чаще наблюдается в среднем возрасте. Она редко развивается в первые 2 десятилетия жизни. Как правило, аритмия возникает во сне или в покое. Хотя синдром Бругада обусловлен аутосомно-доминантным геном, нарушения ритма намного чаще наблюдаются у мужчин.
Ни один из известных антиаритмических препаратов не продемонстрировал способность эффективно предотвращать ФЖ, однако существуют данные об эффективности хинидина при «аритмическом шторме». Единственным методом лечения является имплантация автоматического дефибриллятора. Такое устройство должно быть имплантировано всем пациентам, перенесшим синко-пальное состояние или реанимацию в связи с ФЖ.
Факторы риска при синдроме Бругада
Надежных критериев выявления пациентов с высоким риском, к сожалению, не существует. Исследования в этой области весьма ограничены в связи с относительно малым количеством больных и коротким периодом наблюдения, а также из-за больших различий в частоте зарегистрированных случаев внезапной смерти.
В некоторых работах сообщается о довольно высокой частоте фибрилляции желудочков (ФЖ) у ранее бессимптомных пациентов (8% за 3 года), в то время как другие авторы приводят данные о более низком риске (2% за 5 лет или 0,5% за 30 мес).
Есть мнение о целесообразности стимуляции желудочков при синдроме Бругада I типа. В соответствии с этой позицией имплантация дефибриллятора должна быть рекомендована пациентам, у которых в ходе такого исследования развивается ФЖ, однако в последующих исследованиях это положение не нашло подтверждения.
В качестве возможных факторов риска разными исследователями рассматривались поздние потенциалы желудочков и увеличение продолжительности комплекса QRS, а также нарастание степени выраженности подъема сегмента ST во время теста с физической нагрузкой. Удивительно, но наличие случаев внезапной сердечной смерти в семейном анамнезе, по-видимому, также нельзя считать фактором риска.
Общее мнение заключается в том, что риск меньше у пациентов, у которых отсутствуют спонтанные ЭКГ-признаки синдрома Бругада 1 типа.
Лечение синдрома Бругада
Рутинная имплантация дефибрилляторов бессимптомным пациентам не оправданна в связи с низким риском внезапной смерти, а также хорошо известной и довольно высокой частотой осложнений, связанных с использованием этих устройств, в том числе и при длительном наблюдении за больными. Недавно было предложено назначать в таких случаях хинидин.
Пациентам с синдромом Бругада рекомендуется воздерживаться от приема антиаритмических препаратов I класса (таких как флекаинид) и как можно быстрее начинать лечение любого заболевания, сопровождающегося лихорадкой.
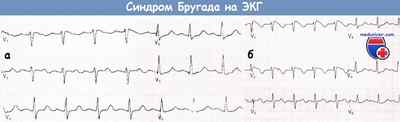
ЭКГ, записанная в грудных отведениях у пациента с подозрением на синдром Бругада (а).
После введения аймалина регистрируется ЭКГ, типичная для синдрома Бругада I типа (б).
Бессимптомный пациент с синдромом Бругада (отведения V1-V3), у которого во время исследования со стимуляцией желудочков развилась ФЖ.
После 8-го навязанного комплекса при частоте стимуляции 120 имп./мин парой преждевременных стимулов инициирована ФЖ.
(Больному был установлен имплантируемый кардиовертер-дефибриллятор (ИКД) и в последующем отмечалось несколько адекватных нанесений электрических разрядов.)
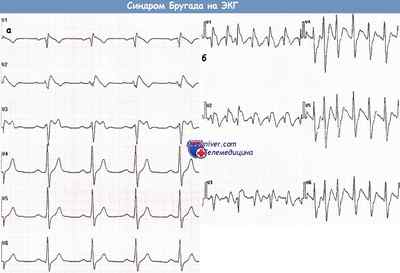
Пациент с синдромом Бругада (а), у которого развилась фибрилляция предсердий (ФП) (б)
http://meduniver.com/Medical/cardiologia/ekg_pri_sindrome_brugada.html
Диагностические признаки синдрома Бругада на ЭКГ
Синдром Бругада — редкое наследственное сердечно-сосудистое расстройство, характеризующееся нарушениями, влияющими на электрические импульсы сердца. Основным симптомом является нерегулярное сердцебиение, без лечения, может привести к внезапной смерти.
Недавние сообщения свидетельствуют о том, что он ответственен за 20% случаев внезапной смерти людей, имеющих сердечно-сосудистые заболевания. Клинический фенотип проявляется во взрослой жизни, чаще встречается у мужчин.
Внезапная смерть может стать первым и единственным проявлением болезни. Бругадский синдром — генетическое заболевание, наследуемое по аутосомно-доминантному признаку. Распространенность — 5 из 10 000 человек.
У нормального сердца четыре камеры. Две верхние камеры известны как предсердия, две нижние камеры — желудочки. Электрические импульсы заставляют сердце биться.
У индивидуумов с синдромом Бругада электрические импульсы между желудочками становятся нескоординированными (фибрилляция желудочков), что приводит к уменьшению кровотока. Снижение кровотока в мозг и сердце приводит к обморокам или внезапной смерти.
Синдром назван испанскими кардиологами Педро Бругадой и Хосепом Бругадой, которые сообщили о нем как о клиническом синдроме в 1992 году. Генетическая основа была установлена Рамоном Бругадой в 1998 году.
Признаки и симптомы
У пострадавшего человека с синдромом Бругады обычно начинают проявляться симптомы в возрасте 40 лет. У людей возникают нерегулярные сердечные сокращения (желудочковые аритмии) или нет явных симптомов (бессимптомно). Неравномерное сердцебиение вызывает затрудненное дыхание, потерю сознания или обморок, внезапную смерть.
Тяжесть симптомов варьируется. Известны триггеры синдрома Бругады, это лихорадка и натриевые блокирующие препараты.
Конкретная презентация синдрома Бругады известна как синдром внезапной ночной смерти (SUNDS). Распространен в Юго-Восточной Азии, встречаются у молодых людей, которые умирают от остановки сердца во время сна без видимой или идентифицируемой причины.
Синдром Бругада вызван мутациями в гене SCN5A, который кодирует ?-субъединицу стробирующего напряжения Nav1.5, сердечного натриевого канала, ответственного за регулирование быстрого тока натрия -INa-. Он вызывает нарушенное функционирование субъединиц натриевых каналов или белков, которые их регулируют. Дисфункция натриевых каналов приводит к локальным закупоркам проводимости в сердце.
В настоящее время сообщалось о более чем 250 мутациях, связанных с BrS, в 18 разных генах (SCN5A, SCN1B, SCN2B, SCN3B, SCN10A, ABCC9, GPD1L, CACNA1C, CACNB2, CACNA2D1, KCND3, KCNE3, KCNE1L -KCNE5-, KCNJ8, HCN4, RANGRF, SLMAP, TRPM4), которые кодируют каналы натрия, калия, кальция или белки, связанные с этими каналами. Несмотря на выявление 18 ассоциированных генов, 65% -70% клинически диагностированных случаев остаются без идентифицируемой генетической причины.
Большая часть мутаций унаследована аутосомно-доминантным образом от родителей детям. Это означает, что для появления болезни необходима только одна копия аномального гена. У большинства людей с заболеванием есть пострадавший родитель. Каждый ребенок пострадавшего человека имеет 50% -ный шанс наследовать генетическую вариацию, независимо от пола.
Первичный ген, который связан с синдромом Бругада, расположен на хромосоме 3, назван геном SCN5A. Приблизительно 15-30% людей с Бругада имеют мутацию гена SCN5A. Ген отвечает за продуцирование белка, который позволяет перемещать атомы натрия в клетки сердечной мышцы через натриевый канал.
Аномалии гена SCN5A изменяют структуру или функцию натриевого канала и приводят к уменьшению содержания натрия в клетках сердца. Уменьшенный натрий приводит к аномальному сердечному ритму, который провоцирует внезапную смерть. Мутации связаны с синдромом QT типа 3 (LQT3), который является одной из форм аномалии сердечного ритма, называемой синдромом Романо-Уорда. Сообщалось, что некоторые семьи имеют родственников с патологией Бругада и LQT3, что указывает на то, что состояния могут быть разными типами одного и того же расстройства.
Распространенность
Синдром Бругада чаще встречается у мужчин (5-8 раз). Встречается во всем мире, но чаще у людей Юго-Восточной Азии, Японии как поккури («внезапная смерть»), Таиланде — Лай Тай («смерть во сне»), широко известен на Филиппинах как бангунгут («стонать во сне»). Согласно медицинской литературе, синдром Бругада составляет 4 — 12 процентов всех внезапных смертей, до 20 процентов всех смертей людей с сердечно-сосудистыми заболеваниями.
Бругада поражает людей любого возраста. Средний возраст внезапной смерти составляет 41 год.
Похожие нарушения
Симптомы следующих расстройств могут быть сходными с симптомами синдрома Бругада. Сравнения полезны для дифференциального диагноза:
Синдром Романо-Уорда
Наследственное сердечное расстройство, характеризующиеся нарушениями, влияющими на электрическую систему сердца. Тяжесть синдрома Романо-Уорда сильно варьируется. У некоторых людей нет явных симптомов; другие развивают аномально повышенные сердечные сокращения (тахиаритмии), приводящие к эпизодам бессознательного (синкопа), остановке сердца, потенциально внезапной смерти.
Синдром Романо-Уорда наследуется как аутосомно-доминантный признак. Один тип синдрома Романо-Уорда, называемый длинным синдромом QT типа 3 (LQT3), вызван аномалиями гена SCN5A; поэтому LQT3 и Бругада могут являться разными типами одного и того же расстройства.

Артериогенная кардиомиопатия (AC)
Редкая форма неишемической кардиомиопатии, при которой нормальная мышечная ткань правого желудочка заменяется жировой тканью. Может развиваться в детстве, но не появляться до 30 — 40 лет. Симптомы AC: нерегулярные сердечные сокращения (аритмии), одышка, опухшие шейные вены, дискомфорт в области живота, обмороки. В некоторых случаях симптомы не проявляются до остановки сердца, внезапной смерти.
Мышечная дистрофия Дюшенна (МДД)
Мышечное расстройство, одно из наиболее частых генетических состояний, затрагивающих 1 из 3500 новорожденных мужского пола во всем мире. Обычно проявляется от трех до шести лет. МДД характеризуется слабостью, отмиранием (атрофией) мышц тазовой области с последующим вовлечением мышц плеча. По мере прогрессирования болезни мышечная слабость и атрофия распространяются по всем мышцам тела. Болезнь прогрессирует, большинство страдающих людей, нуждаются в инвалидной коляске в подростковые годы.
Развиваются серьезные, опасные для жизни осложнения — болезни сердечной мышцы (кардиомиопатия), затрудненное дыхание. DMD вызван изменениями (мутациями) гена DMD на Х-хромосоме. Ген регулирует продуцирование белка, называемого дистрофином, который играет важную роль в поддержании структуры внутренней стороны мембраны клеток скелетной и сердечной мышц.
Дополнительные расстройства имеющие аналогичные аномалии сердечного ритма: острый миокардит, острая легочная тромбоэмболия, ишемия правого желудочка или инфаркт, дефицит тиамина, гиперкальциемия, гиперкалиемия.
Диагностика
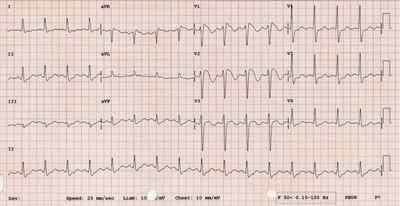
Диагностика синдрома Бругада основана на тщательной клинической оценке, полной медицинской и семейной истории внезапной сердечной смерти, специализированный тест, известный как электрокардиограмма (ЭКГ), регистрирующий электрическую активность сердца. Врачи используют специальные препараты (блокаторы натриевых каналов), провоцирующие характерные особенности ЭКГ синдрома Бругада.
Молекулярно-генетическое (ДНК) тестирование для мутаций во всех генах проводится для подтверждения диагноза. Только 30-35% пораженных людей имеют идентифицируемую мутацию гена после всестороннего генетического теста. Последовательный анализ гена SCN5A является первым шагом молекулярно-генетического диагноза, поскольку мутации этого гена наиболее распространенная причина синдрома Бругада (около 25%).
Постановка диагноза
Диагноз может быть сложным, так как ЭКГ человека с синдромом Бругада может быть совершенно нормальной. В этих случаях, диагноз устанавливается путем повторения ЭКГ с помощью введения лекарственного средства, которое выявляет специфические аномалии, наблюдаемые при этом состоянии (например, вызов Ajmaline или Flecanide). Или путем тестирования ДНК идентифицируя определенную мутацию гена.
Изменения ЭКГ могут быть временными при Бругада, но провоцируются несколькими факторами:
- лихорадка
- ишемия
- Блокаторы натриевых каналов, например, Flecainide, Propafenone
- Блокаторы каналов кальция
- Альфа-агонисты
- Бета-блокаторы
- Нитраты
- Холинергическая стимуляция
- Алкоголь
- Гипокалиемия
- гипотермия
Диагностические критерии
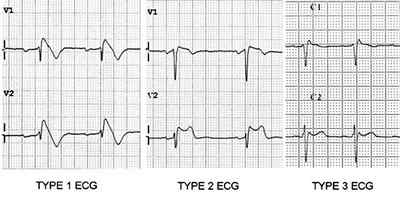
Тип 1 (возвышение сегмента Coved ST; 2 мм; 1 из V1-V3, за которым следует отрицательная T-волна) — единственная аномалия ЭКГ, которая потенциально диагностируется. Упоминается как знак Бругада.
Знак Бругада
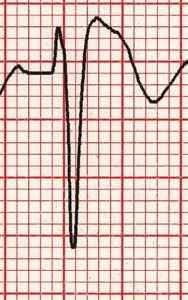
Эта аномалия ЭКГ должна быть связана с одним из клинических критериев для постановки диагноза:
- Документированная фибрилляция желудочков (VF) или полиморфная желудочковая тахикардия (VT).
- Семейная история внезапной сердечной смерти в возрасте 45 лет.
- ЭКГ с кубическим типом у членов семьи.
- Индуцируемость VT с запрограммированной электрической стимуляцией.
- Обморок.
- Апное.
Два других типа не являются диагностическими, требуют дальнейшего изучения
- Brugada Type 2: имеет 2 мм седлообразной формы ST.
- Brugada type 3: может быть морфологией типа 1 или 2, но с lt;2 мм высоты сегмента ST.
Клинические испытания
Электрокардиография болезни рекомендуется для определения степени заболевания. Электрофизиологическое исследование используется для оценки риска внезапной сердечной смерти.
Нет лечения синдрома Бругада. Лица имеющие высокий риск фибрилляции желудочков лечатся имплантируемым дефибриллятором кардиовертера (ICD). Это устройство автоматически обнаруживает аномальное сердцебиение и выборочно подает электрический импульс в сердце, восстанавливая нормальный ритм.
Изопротеренол – антиаритмический препарат, используется для эффективного ответа на электрические бури (нестабильные желудочковые аритмии). Рекомендации по лечению бессимптомных индивидуумов противоречивы. Возможные виды лечения включают: наблюдение до появления симптомов, хотя первым симптомом — внезапная сердечная смерть или использование семейной истории, электрофизиологические исследования.
Генетическая консультация рекомендуется для пострадавших лиц и их семей. Другое лечение симптоматическое, поддерживающее.
Например, агрессивное лечение лихорадки лекарствами для снижения температуры (парацетамол), поскольку лихорадка любого происхождения может вызвать опасные аритмии.
Следует избегать определенных лекарств, современные рекомендации можно найти на сайте.
Специалист определяет риск развития угрожающих жизни аритмий, может быть рекомендовано имплантируемый дефибриллятор (ICD). ICD рекомендован, когда пациент уже перенес опасную аритмию. Некоторые препараты исследуются как лечение нарушений ритма. Как только человеку поставлен диагноз синдрома Бругада, все родственники первой линии должны быть проверены.
Важно знать, что многие люди, у которых диагностировано поражение живут долго и счастливо.
- существует только один тип синдрома Бругада.
- Диагноз зависит от характерного ЭКГ, клинических критериев.
- Знак Бругада в изоляции имеет сомнительное значение.